Case Report
Endocrine abnormalities in two siblings with Rothmund Thomson Syndrome

Nagehan Aslan1* and Ozgur Pirgon2
1Department of Pediatrics, Division of Pediatric Intensive Care, Cukurova University, Faculty of Medicine, Adana, Turkey
2Professor, Department of Pediatrics, Division of Pediatric Endocrinology, S. Demirel University, Faculty of Medicine, Isparta, Turkey
*Address for Correspondence: Nagehan Aslan, MD, Department of Pediatrics, Cukurova University, Faculty of Medicine, Adana, Turkey, Tel: +05055499986; Email: [email protected]
Dates: Submitted: 03 October 2018; Approved: 10 October 2018; Published: 11 October 2018
How to cite this article: Aslan N, Pirgon O. Endocrine abnormalities in two siblings with Rothmund Thomson Syndrome. Ann Clin Endocrinol Metabol. 2018; 2: 041-045. DOI: 10.29328/journal.acem.1001010
Copyright License: © 2018 Aslan N, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Rothmund-thomson syndrome; Cryptorchidism; Endocrine

Abstract
Rothmund-Thomson syndrome is a rare autosomal recessive disorder characterized by poikiloderma (skin atrophy, telangiectasia, hyper- and hypopigmentation), congenital skeletal abnormalities, short stature, premature aging, and increased risk of malignant disease. Two siblings with Rothmund-Thomson Syndrome showed the following characteristic features: severe growth failure, dystrophic nails, absent eyelashes/eyebrows, small hands, clinodactyly, microdontia and congential poikiloderma. In addition, delayed sexual development with cryptorchidism in the male and Hashimato thyroiditis in the female patient were detected. These cases are presented here because of these endocrine patterns, with the aim of drawing attention to the invisible aspects of Rothmund-Thomson syndrome.
Introduction
Rothmund-Thomson syndrome (RTS) or poikiloderma congenitale, is an extremely rare autosomal recessive genetic condition which exhibits some dermatological, craniofacial, ophthalmological and central nervous system abnormalities [1]. Depending upon genetic and phenotypic heterogenity, two clinical forms may occur. RTS Type 1 has poikiloderma, hypogonadism, juvenile cataract and no identified gene mutation. Type 2 is seen with poikiloderma and skeletal abnormalities and is caused by homozygous or compound heterozygous mutation RECQL4 helicase gene mapped to chromosome 8q24. RTS features a DNA damage repair defect and presents in infancy with specific facial rashes and heterogeneous clinical characteristics such as short stature, hair and nail abnormalities, photosensitivity, juvenile cataracts and skeletal changes with a predisposition to skin cancers and osteosarcoma [2]. In addition, more rare symptoms are endocrine disorders such as hypogonadism, amenorrhoea, and a lack of secondary sex characterstics. In this paper, the endocrinological aspects of RTS in two siblings of different sex are presented and RTS is discussed in the light of the literature.
Description of Cases
Patient 1
A 14-year old female presented with absent eyelashes/eyebrows and poikiloderma involving the face, extremities and buttocks and growth retardation. She was the first child born to non-consanguinous parents after normal gestation and her brother had similar complaints. Since 3 months of age, she would develop facial erythema and edema after sun exposure. Her height was 139.7 cm (<3 percentile, SDS:-2.76) and weight was 30.7 kg (<3 percentile, SDS: -2.04). Bone and chronological age were consistent and her mental capacity was normal on the Denver Development Screening Test. She had microdontia, tooth development delay, small hands, clinodactyly in the fifth fingers of both hands, and strabismus in addition to palmoplantar hyperceratosis and dystrophic nails (Figure 1a,b). Dermatological consultation and skin biopsy showed features of poikiloderma. The white blood cell count was 2900/mm3, hemoglobin 13.2gr/dL, thrombocyte count 290.000/mm3, and serum biochemistry, urine analysis and imaging studies were normal. She was receiving treatment for hypothyroidism due to Hashimato thyroiditis diagnosed at the age of 9 years. Anti-thyroid peroxidase level was 530 U/mL, and thyroid ultrasonography showed thyroid glands with heterogeneous echogenicity. The thyroid hormone levels were within the normal range with thyroxine treatment. Genetic studies were performed for the diagnosis of RTS, however, RECQL4 mutation was not detected.
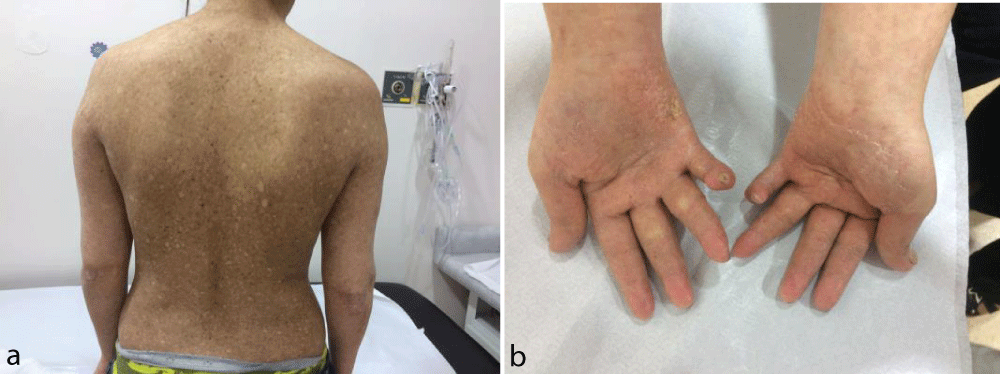
Figure 1a,b:
Patient 2
A 12-year old male presented with poikiloderma and growth retardation. He was the second child of the same family born after normal gestation. Poikiloderma had developed over the years involving the face, extremities and buttocks. The history of facial erythema and edema after sun exposure and leukopenia was similar to Case no 1. He had undergone cryptorchidism surgery at the age of 5 years and had been monitored in respect of leukopenia since the age of 2 years. Bone marrow aspiration was performed but it had normal maturation. His height was 131.3 cm (<3 percentile, SDS: -2.71) and weight was 27.4 kg (<3 percentile, SDS: -1.48). Bone and chronological age were consistent and his mental capacity was normal on the Denver Development Screening Test. He had microdontia, palmoplantar hyperceratosis and dystrophic nails on the hands and feet in addition to the widespread poikiloderma (Figure 1c,d). Testis volume was 2 mL and penis size was 3 cm. The results of the ophthalmological, neurological and scalp hair examinations were normal. White blood cell count was 2600/mm3, hemoglobin 12.1 gr/dL, thrombocyte count 160.000/mm3, and serum biochemistry, urine analysis and imaging studies were normal. Dermatological counselling was given. A skin biopsy showed features of poikiloderma. For the diagnosis of RTS, genetic studies were performed, however, RECQL4 mutation was not detected. In the 3-year follow-up period in our clinic, no increase was determined in the volume of the testes. A low plasma testosterone level, and elevated LH and FSH levels were the main endocrine anomalies in this patient. Table 1 and Table 2 show the clinical characteristics and laboratory findings of these patients. Ethical approval was received from Suleyman Demirel University ethics committee.
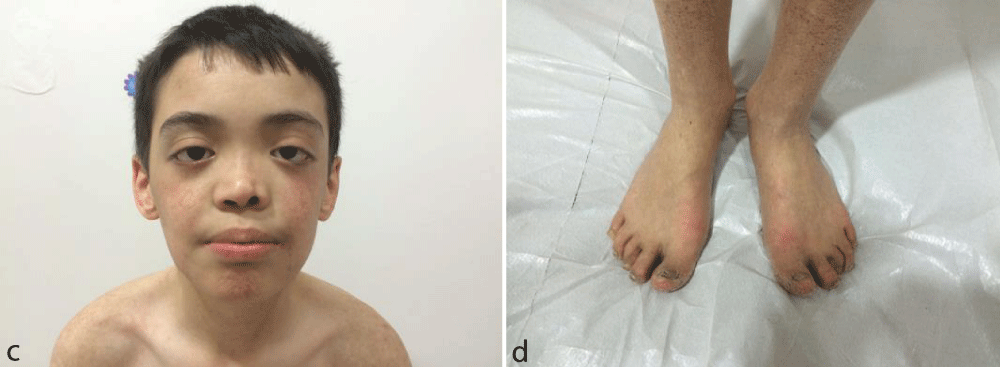
Figure 1c,d:
| Table 1: Clinical characteristics of patients | ||
| Patient 1 | Patient 2 | |
| Age | 14 | 12 |
| Gender | female | male |
| Growth retardation | + | + |
| Skeletal abnormalities | ||
| Clinodactyly | + | - |
| Small hands | + | - |
| Skin abnormalities | ||
| Poikiloderma | + | + |
| Dystrophic nails | + | + |
| Absent eyelashes | + | - |
| Ophthalmic abnormalities | ||
| Juvenile cataract | - | - |
| Strabismus | + | - |
| Endocrine abnormalities | ||
| Hypogonadism | - | + |
| Hypothyroidism | + | - |
| Dental abnormalities | ||
| Tooth delay | + | + |
| Microdontia | + | + |
| Hematological abnormalities | ||
| Microcytic anemia | - | - |
| Leukopenia | + | + |
| Table 2: Laboratory findings of the patients. | ||
| Patient 1 | Patient 2 | |
| White blood cell count (mm3) | 2900 | 2600 |
| Free T4 (ng/dL) | 0.53 | 0.82 |
| TSH (mIU/mL) | >100 | 2.5 |
| Anti-TPO(IU/mL) | >1000 | 10 |
| FSH (mIU/mL) | 6.5 | 72 |
| LH (mIU/mL) | 3.4 | 6.4 |
| Estradiol | 45 | - |
| Total testosterone (ng/dL) | - | 57 |
Outcome and follow-up of the siblings
In the second year after diagnosis of Case no 1, secondary sexual characteristics developed and menarche began. Three years after diagnosis she was admitted with an unhealed traumatic wound, 3x2 cm in size in lower part of her leg. This wound had been present for over 1 month and over this period and she had received no treatment. As a result of dermatological and plastic reconstructive surgery consultation, an incisional biopsy was performed but no neoplastic changes were observed. Local therapy of topical medication was applied and debridement was not necessary.
In Case no 2, hypergonadotropic hypogonadism with small testes and high basal LH and FSH levels as well as increased LH and FSH response to LH-RH stimulating test were found. Stimulation tests and pregnyl treatment were applied but no response was obtained.
Discussion
Both cases had poikiloderma and growth retardation, and the endocrine abnormalities were particularly striking. Primary hypogonadism was confirmed in second case by endocrinological investigations. Some individuals with RTS may demonstrate hypogonadism [3,4], a condition characterized by deficient activity of the ovaries in females or testes in males [5,6]. Affected females may experience irregular menstruation, while both affected males and females may have delayed sexual development. In individuals with hypogonadism, fertility may be reduced, although some patients have children. Hypothyroidism has been suggested by some authors as a possible cause of a puffy face in some cases. Hypoparathyroidism has been previously defined in RTS [6]. Case no 1 had regular menarche and hypothyroidism, while Case no 2 had hypogonadism. A literature scan revealed a case report of three siblings with RTS at pediatric age with hypogonadism [7]. However, no other cases were found of hypothyroidism, as in the current case. To the best of our knowledge, these two presented cases differ from the others in this respect with these endocrine abnormalities.
Leg ulcer is rare in RTS. There have been a few case reports in literature in the adult age group. No pediatric reports could be found in this regard and our female patient seems to be the first reported pediatric RTS case with a leg ulcer. The incidence of dental anomalies (microdontia, hyperdontia, early caries) has been estimated as 27% - 59% [8]. The current cases had defective dentition. Mental retardation is a rare condition which is seen in approximately 10% of patients [9]. The current cases had normal mental development.
Gastrointestinal (eosophageal stenosis, chronic emesis, diarrhea), and hematological (microcytic hypochromic anemia, leukopenia) abnormalities occur infrequently [2]. Both of the current cases were examined for leukopenia. There is also known to be an increased risk for skin and extracutaneous malignancies such as osteosarcoma, myelodysplastic syndrome, Hodgkin’s disease, squamous cell carcinoma, and basal cell carcinoma [10].
This syndrome is caused by a DNA damage repair defect. The differential diagnosis should include other causes of childhood poikiloderma (dyskeratosis congenita, Kindler syndrome and poikiloderma with neutropaenia), and other rare genodermatoses with prominent telangiectasias (Bloom syndrome, Werner syndrome and ataxia-telangiectasia).
Germline mutations in RECQL4 are found in the majority of RTS patients and RECQL4 is the only gene that is currently known to be associated with RTS. In literature, RECQL4 mutations have been reported to be detected in 60-65% of patients with a clinical diagnosis of RTS [2]. The presence of mutations in RECQL4 can confirm a diagnosis of RTS, although the absence of mutations in RECQL4 does not necessarily mean that the patient does not have RTS, as one-third of patients clinically diagnosed with RTS do not have mutations in this gene. Similarly, the RECQL4 mutation was not detected in the current cases.
In conclusion, it can be said that RTS is a rare clinical entity and endocrine abnormalities in RTS are even more rare. The cases presented here had endocrine disorders in addition to skin, skeletal, ophthalmological, dental and hematological abnormalities and are therefore cases of interest. Similarly, leg ulcer is a rare and important clinical presentation of RTS because of the increased risk of skin malignancy. These cases can be considered interesting in these aspects and this report will make a contribution to literature.
Ethics Committee Approval: The ethics committee approval has obtained from the Suleyman Demirel University Ethics committee.
Informed Consent: Written informed consent was obtained.
References
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999; 22: 82-84. Ref.: https://goo.gl/AdkJ5R
- Bloch B, Stauffer H. Poikiloderma-like changes in connection with underdevelopment of the sexual glands and dystrophia adiposogenitalis. Archives of Dermatology and Syphilology. 1929; 19: 22-34. Ref.: https://goo.gl/swFK9A
- Vennos EM, Collins M, James WD. Rothmund-Thomson syndrome: review of the world literature. J Am Acad Dermatol. 1992; 27: 750-759. Ref.: https://goo.gl/oq8ij4
- Werder EA, Mürset G, Illig R, Prader A. Hypogonadism and parathyroid adenoma in congenital poikiloderma (Rothmund-Thomson syndrome). Clin Endocrinol. 1975; 4: 75-82. Ref.: https://goo.gl/hnWH3Y
- Guyda H, MacLeod P, Colle E. Endocrine Aspects of the Rothmund-Thomson syndrome. Pediatric Research. 1975; 9: 689. Ref.: https://goo.gl/WZYpX3
- Haytaç MC, Oztunç H, Mete UO, Kaya M. Rothmund-Thomson syndrome: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 94: 479-484. Ref.: https://goo.gl/eankRi
- Polese L, Merigliano S, Mungo B, Pennelli G, Norberto L. Report on a case of Rothmund-Thomson syndrome associated with esophageal stenosis. Dis Esophagus. 2011; 24: 41-44. Ref.: https://goo.gl/Dq6L2J
- Stinco G, Governatori G, Mattighello P, Patrone P. Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. J Dermatol. 2008; 35: 154-161. Ref.: https://goo.gl/ctTqnt
- Sexton GB. Thornson’s syndrome (poikiloderma congenitale). Canadian Medical Association Journal. 1954; 70: 662.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet Journal of Rare Diseases. 2010; 5: 2. Ref.: https://goo.gl/SiQ8Ne